Understanding active site chemistry of hydrogenase enzymes
A wide range of micro-organisms express hydrogenases - highly active
enzymes for oxidation of H2, or reduction of
H+ to produce H2. The efficiency of
H2/H+ interconversion by hydrogenases is providing inspiration for development of new catalysts for fuel cells or
clean H2 production built from abundant metals, but this requires an intimate knowledge of how H2 is activated at hydrogenase active sites. The Vincent group are using a combination of electrochemical and spectroscopic methods to understand hydrogenase active site chemistry.
Direct electrochemical methods, in which
hydrogenases are adsorbed as a sub-monolayer film onto a graphite electrode,
provide precise control over active site chemistry. In this approach called (protein
film electrochemistry), adsorbed enzyme molecules exchange electrons
directly with the electrode so that no electron-transfer mediators are
required. The current at the electrode reports on the catalytic activity of the adsorbed
hydrogenase at each applied potential. Protein film electrochemistry has provided much insight into the
chemistry of hydrogenases.
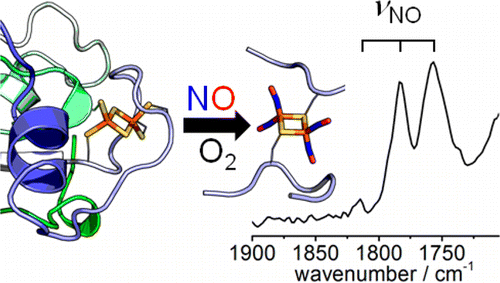
We are using a range of coupled electrochemical and infrared spectroscopic approaches to reveal structural details of enzyme active site chemistry during catalytic turnover. The active sites of
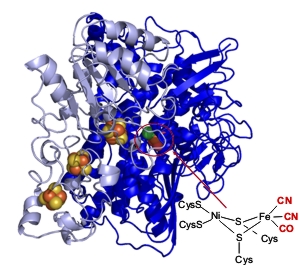
hydrogenases incorporate ligands that are unusual in biology:
carbon monoxide (CO) and cyanide (CN-). These
ligands provide a useful spectroscopic handle for studying structural
changes at the metal centre because vibrational transitions associated
with CO and CN- give rise to relatively intense
absorption bands in the infrared.
Applications of hydrogenases in Industrial Biotechnology
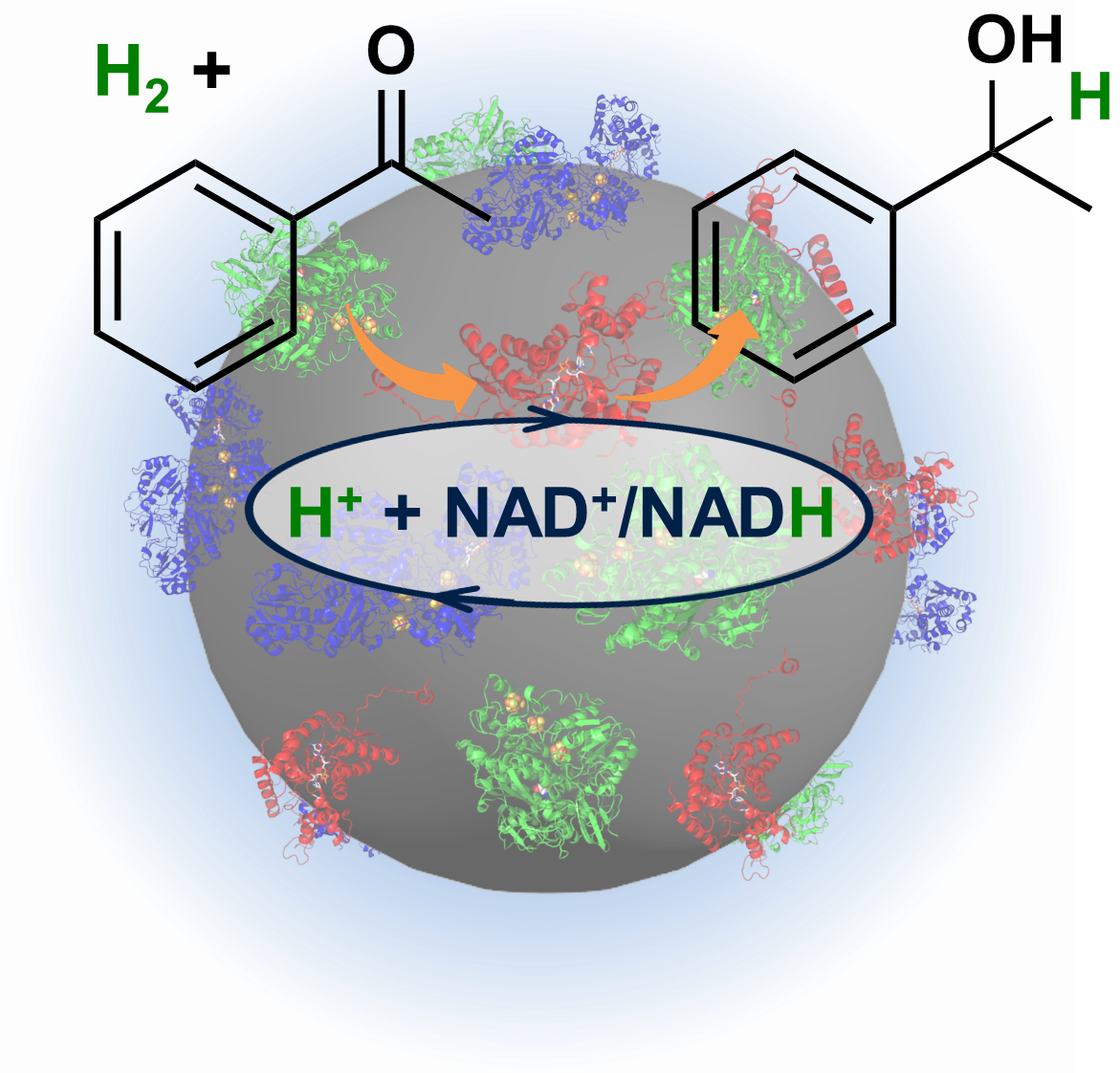
We have developed a series of applications of hydrogenases for H2-driven biocatalysis. One of these, the HydRegen catalyst system, uses enzymes for both H2-driven NADH recycling and a selective NADH-dependent biotransformation 'plugged' into carbon particles. We have also developed systems for H2-driven recycling of reduced flavins with applications in catalysis by ene reductases and nitro reductases. We are working on a range of chemo-bio catalyst systems. For these projects we have close industrial collaborations as well as many academic collaborators.
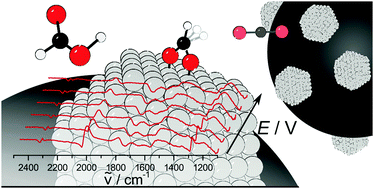
Spectroelectrochemistry in the study of energy-related catalysts
We have applied IR spectroelectrochemistry to examine surface chemistry and poisoning of supported nanoparticles of platinum and other precious
metals in fuel cell electrocatalysis, under fuel flow conditions. We are working with academic collaborators on a range of other mechanistic projects in energy catalysis.